We are big believers in automated test suites to ensure that software tools and workflows produce the results we expect them to, both now as well as after any changes we make in the future. This is particularly important in a project like ours, where there are many moving parts, from software code in different languages and OWL ontologies we develop to 3rd party tools and libraries for RDF and OWL, that all contribute to the end result in ways that would be exceedingly time consuming to trace and test manually whenever something changes. How can we convince ourselves (let alone others) that a change to an OWL data model, template, or a piece of code that generates an OWL ontology according to the model and template, does not result in phyloreferences that produce incorrect results when applied to a phylogeny?
Enter automated test suites. These are tools that test other pieces of software, usually by providing them with valid and invalid inputs to make sure the software being tested responds correctly to all of them. Creating a test suite will ensure that anybody, including ourselves, can quickly check to make sure that all of the tested pieces of software are working together to give the right answer.
We already have some basic tests for our phylo2owl tool, which converts a phylogeny into an OWL ontology. One makes sure that the tool executes without error. Another set of tests checks that an example phylogeny we’ve included in our repository (thanks for the liberal licensing, Mullins et al., 2012!) produces valid RDF/XML that is identical to a copy of expected result we’ve archived in our repository. However, this is both limited and naïve. We’ll know that something is up if the file it produces changed – but a set of axioms can often be written to a file in ways that produces different sequences of bytes, yet represent the exact same or logically equivalent axioms. Figuring out by hand in such a situation what changed, and whether the change is neutral or not, can be time-consuming even for only a handful of input files, let alone many. We’re going to need something more powerful if we want to be able to check hundreds or even thousands of different phyloreferences every time we make changes.
To accomplish this, we’re investigating a set of software tools that can detect the presence or absence of expected (or unexpected) relationships within an RDF document. These include:
-
Shape Expressions (ShEx) come with their own compact syntax for describing the “shape” of RDF data. It has Python, Scala and Javascript libraries.
-
SPARQL Inferencing Notation (SPIN) is a representation of SPARQL in RDF, allowing queries and their expected values to be expressed as RDF. SPIN is maintained by TopBraid, who have produced an open source Java library.
-
Shapes Constraint Language (SHACL) is a W3C working draft similar to ShEx, but expressed completely in RDF. This means that a shape’s description directly resembles the data it describes, and that data and shapes can be provided in the same file. This page has a nice comparison of the different syntax used by ShEx and SHACL, while this presentation describes the deeper differences between the two.
These tools would help us with two main goals:
-
To confirm the basic relationships we’d expect to see: for example, we might expect every Node in a phylogeny will be related phylogenetically to at least one other node in the phylogeny.
-
For a particular phylogeny, we could identify aspects of the structure we would expect to find: for example, a particular phylogeny might show one node being ancestral to another. If the nodes in the OWL ontology don’t have the same relationship, then something is wrong.
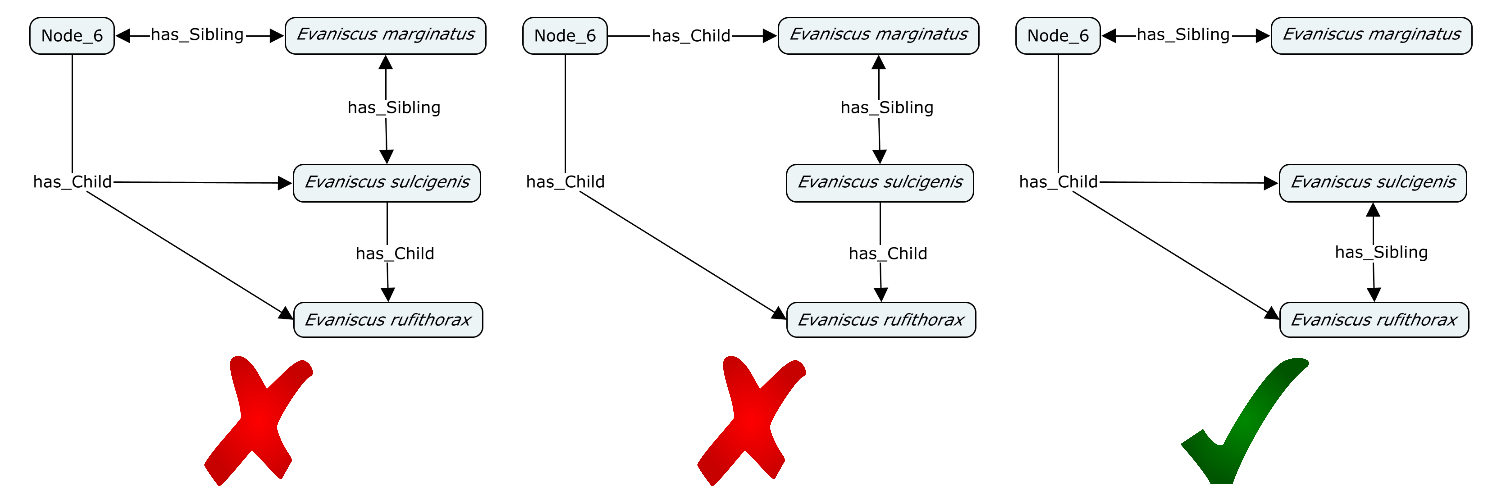
In terms of basic relationships, the most basic test is that every phylogeny should contain at least one Node (CDAO:0000140). If it has more than one Node, then every Node should have either a has_Child or has_Sibling with at least one other Node. Furthermore, no node should be a child of itself.
In terms of specifics, I’m going to look at encoding lineage lines (for example, Evaniscus tibialis is a child of Node_7, which is a child of Node_3, which is a child of Node_2, which is a child of Node_1). If I can come up with a compact representation of these, it may be a neat way to make sure that ontologies are being converted correctly.